 Q-Chem:
Facilitating Worldwide Scientific Breakthroughs
Q-Chem:
Facilitating Worldwide Scientific Breakthroughs
Q-Chem is a robust software
platform with an extensive set of features. Whether you want to study
spin-orbit coupling effects in a single-molecule magnet, run
high-throughput calculations on small organic molecules, study an
enzyme using QM/MM, or something entirely different, our software
package offers a wide range of solutions for a variety of applications.
Check out our available features and see how Q-Chem can help you
achieve your research goals!
|
|
 |
 |
 |
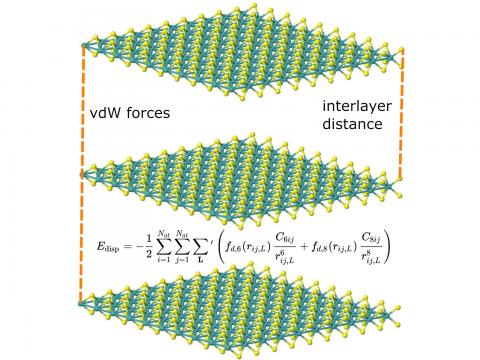
Q-Chem supports LDA, GGA, and meta-GGA functionals, as well as hybrid,
range-separated hybrid, and double hybrid versions of both GGAs and
meta-GGAs. Single-point energies, geometry optimizations, vibrational
frequency calculations, and many other properties can be evaluated for
ground states, and for excited states via time-dependent DFT. |
We are pleased to present the seventh major release of the Q-Chem ab inito quantum chemistry software package, Q-Chem 6.2! Our latest release has many improvements and new features, including:
- Natural Auger Orbitals for Auger decay, ICD, and related processes for CVS-EOM methods (Nayanthara K. Jayadev, Anna I. Krylov)
- ACP-EOMIP-CCSD for partial Auger decay widths (Florian Matz, Thomas Jagau)
- EOM-CCSDT for EE and SF states (Manisha, Prashant Uday Manohar)
- Dipole filtering for TDKS (John Herbert, Avik Kumar Ojha)
- DFT/CIS semi-empirical method, including a new parameterization for X-ray spectroscopy (Aniket Mandal, John Herbert)
- Generalization of 1C-NOCIS to two-electron open-shell singlets (Juanes Arias-Martinez, Hamlin Wu, Martin Head-Gordon)
- Atomic multipole moment calculation using IAOs (Alexandra McIsaac, Abdulrahman Aldossary, Martin Head-Gordon)
- RT-NEO, RT-NEO-Ehrenfest, BO-RT-NEO-Ehrenfest, and RT-NEO-Ehrenfest-QM/MM (Tao E. Li, Mathew Chow, and Sharon Hammes-Schiffer)
- NEO multistate DFT (NEO-MSDFT) (Joseph Dickinson, Qi Yu, and Sharon Hammes-Schiffer)
- SCS-RIMP2 and SOS-OOMP2 for NEO methods (Jonathan Fetherolf and Sharon Hammes-Schiffer)
For a complete list of new features, bugfixes, and improvements, please see the Q-Chem 6.2 release log.
|
Q-Cloud (coming in January of 2025) provides a fast, easy way to run Q-Chem calculations on Amazon's AWS infrastructure,
providing improved flexibility and fast turn-around time on jobs while
reducing compute costs. The benefits of cloud computing include:
- Flexibility. The
number of nodes on your Q-Cloud cluster automatically scales on demand,
based on what is actually required to run your jobs. Whether you need
to run running a hundred jobs at once, or just a handful, you'll only
pay for the hardware you use.
- Sustainability. According to both Microsoft and AWS,
cloud computing options can be 22—93% more energy efficient than
traditional on-premises infrastructure, depending on the specific setup.
- Reduced infrastructure costs. Never
spend valuable research time troubleshooting faulty hardware again! AWS
maintains their own cloud computing infrastructure, so you don't have
to, and the Q-Cloud installation process is simple and quick, making it
easy to get up and running.
- Fixed-Cost Software-as-a-Service (SaaS) Payment Model. The
cost of all standard Q-Cloud licenses is one single monthly or annual
payment, and payment for AWS resources scales with use. Additionally,
you will always have access to the most recent version of our software.
|
Q-Chem offers
state-of-the-art tools for treating electron correlation effects, such
as Møller-Plesset perturbation theory and coupled-cluster
theory. For systems with strong correlation, Q-Chem offers specialty
treatments including CASSCF, coupled-cluster valence bond theory,
selected CI, RAS-CI, spin-flip, and variational 2-RDM methods.
|
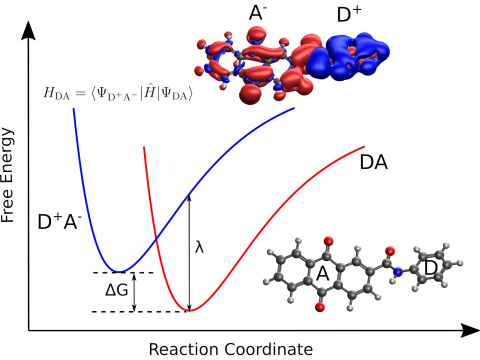
Q-Chem provides a diverse
set of methods for studying electronically excited
states: CIS, TD-DFT, NOCI, EOM-CC, and ADC. Specialty flavors of
these methods cover many types of electronic structure, making it
possible to simulate spectroscopic features, charge and energy
transfer, and non-adiabatic dynamics. Additionally,
our wavefunction analysis module can be used to provide
further insight into excited states.
|
|
|
 |
 |
 |
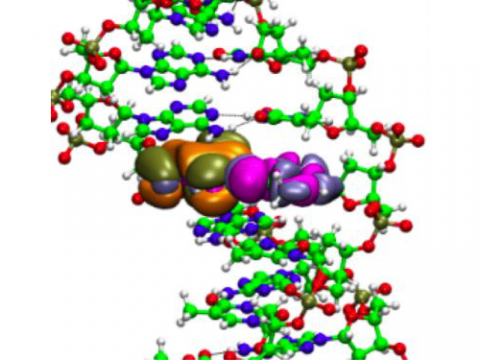
The
Q-Chem package offers a variety of solutions for modeling solvated
systems, ranging from implicit solvent models, such as SM8, COSMO, and
C-PCM, to the effective fragment potential method, which can be used to
capture explicit solvent effects. Additionally, Q-Chem includes several
different embedding approaches, including QM/MM and density embedding,
as well as interfaces to CHARMM and GROMACS. |
|
|
| Q-Chem
offers a variety of tools for modeling different types of spectra. Our
capabilities include IR and Raman spectroscopy, UV-vis
spectroscopy, X-ray spectroscopy, photoelectron spectroscopy, NMR
spectroscopy, and nonlinear spectroscopy (such as two-photon
absorption). Spectroscopic features can be studied using
many different levels of theory, ranging from
TDDFT to EOM-CC and ADC methods. |
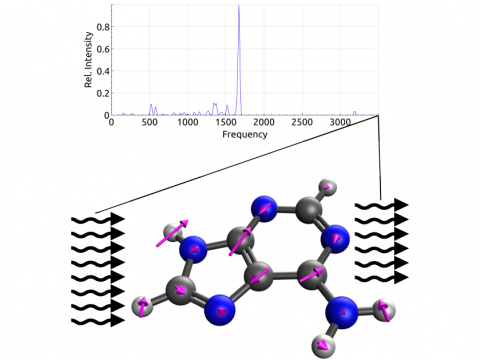
Vibrational Spectroscopy
|
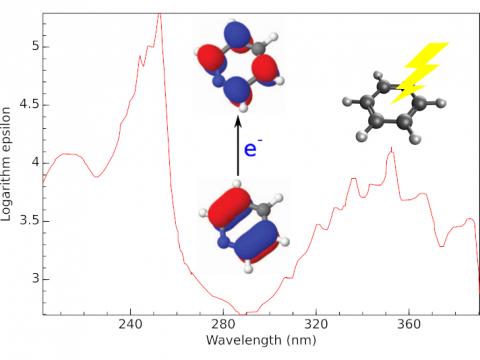
Electronic Spectroscopy
|
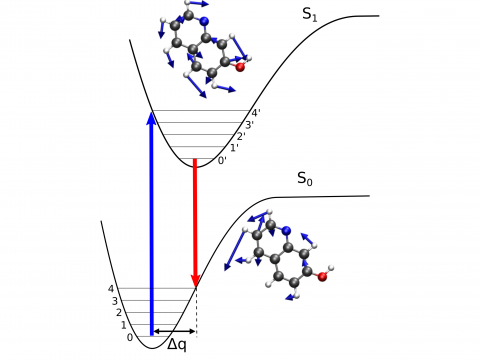
Vibronic Spectroscopy
|
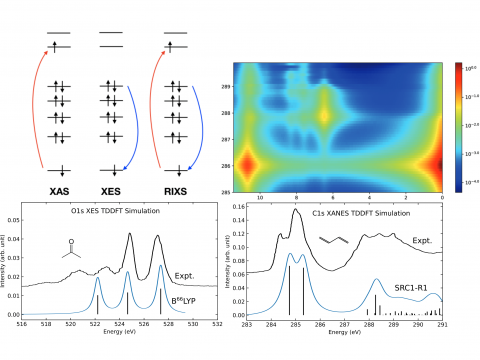
X-ray Spectroscopy
|
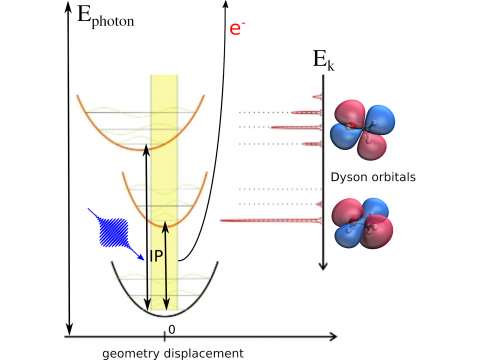
Photoelectron Spectroscopy
|
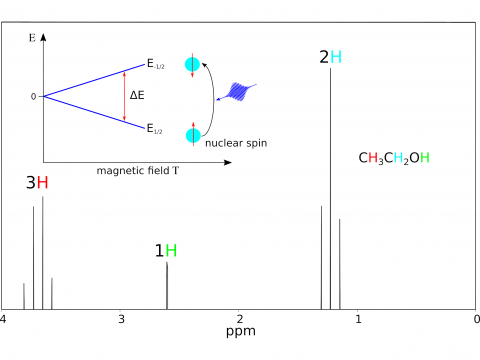
Magnetic Spectroscopy
|
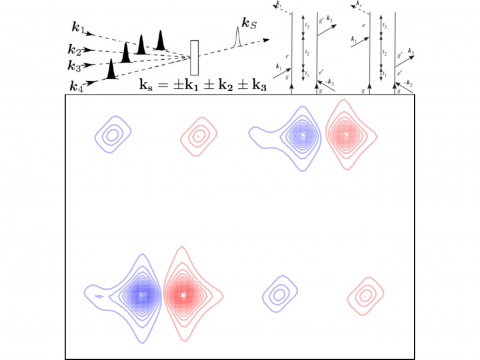
Nonlinear Spectroscopy
|
|
|
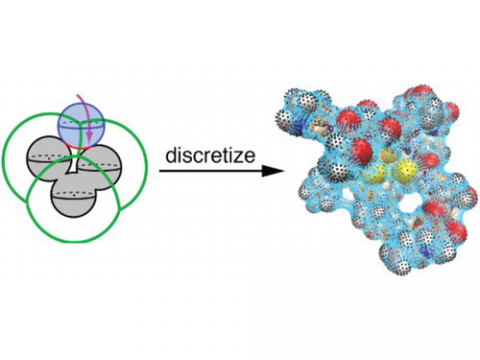
Energy
decomposition analysis based on absolutely localized molecular orbitals
provides a breakdown of the total interaction energy into meaningful
physical terms, providing insights into the nature of intermolecular
and bonded interactions. Symmetry-adapted perturbation theory (SAPT)
and an extended many-body version thereof (XSAPT) are also available
for computing and analyzing intermolecular interactions.
|
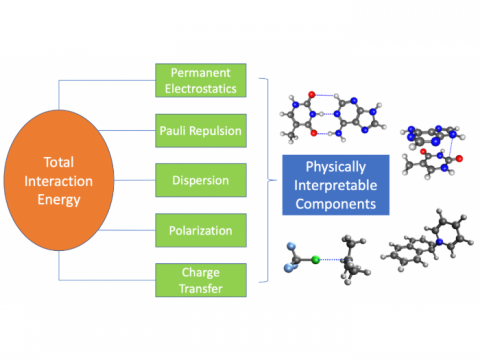
Energy Decomposition Analysis |
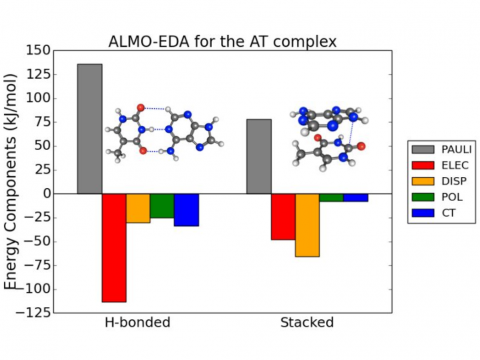
ALMO-EDA for the AT Complex |
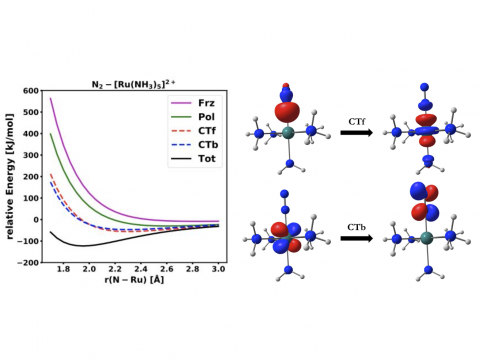
EDA for a Ru Complex |
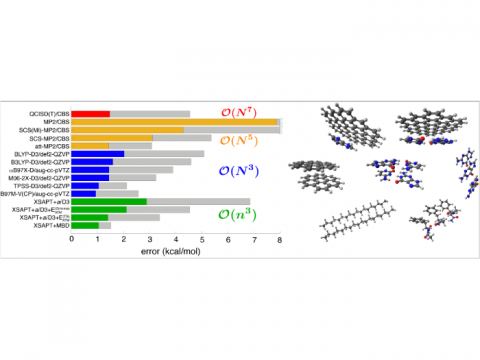
Comparison of Errors |
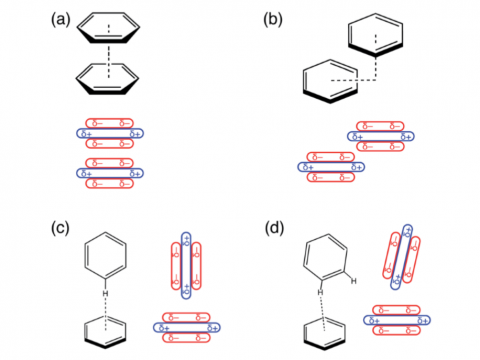
SAPT |
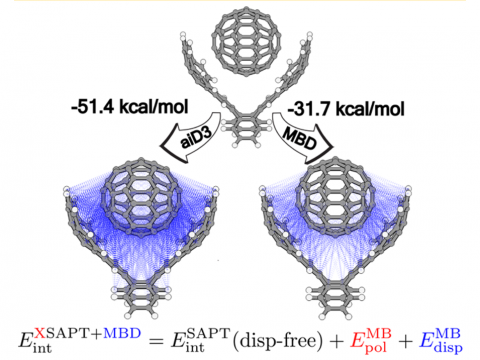
XSAPT |
Q-Chem provides methods
for geometry optimization, potential energy surface scans, transition
state searches, and intrinsic reaction coordinate following, making it
ideal for studies of chemical reactivity, thermochemistry, and chemical
kinetics.
|
|
Q-Chem can perform ab initio molecular
dynamics (AIMD), including both NVE and NVT thermal
samplings, as well as quasi-classical molecular dynamics
(QMD). These approaches can be used to produce vibrational spectra
and ab initio path
integrals. We also include an implementation of Tully's
fewest-switches surface hopping (FSSH) approach to
effectively handle non-adiabatic systems.
|
 |
 |
- Fully
integrated
graphic interface including molecular builder, input generator,
contextual help and visualization toolkit;
- Dispersion-corrected
and double hybrid DFT functionals;
- Faster
algorithms
for DFT, HF, and coupled-cluster calculations;
- Structures
and
vibrations of excited states with TD-DFT;
- Methods for
mapping
complicated potential energy surfaces;
- Efficient
valence space
models for strong correlation;
- More
choices
for
excited states, solvation, and charge-transfer;
- Effective
Fragment
Potential and QM/MM for large systems;
- For
a complete
list of new features, click
here.
|
IQmol:
Q-Chem Graphical User Interface
The smart choice in molecular visualization software!
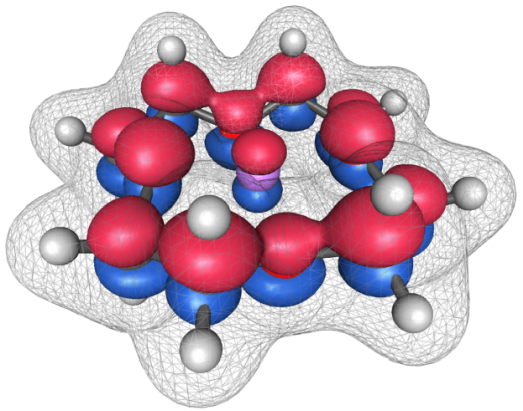
The
above image, generated in IQmol, shows the lowest unoccupied molecular
orbital (LUMO) for the 12-crown-4 ether complexed with a lithium
cation. The mesh superimposed on the system shows the total density.
|
|