Dragon 6
Main Features
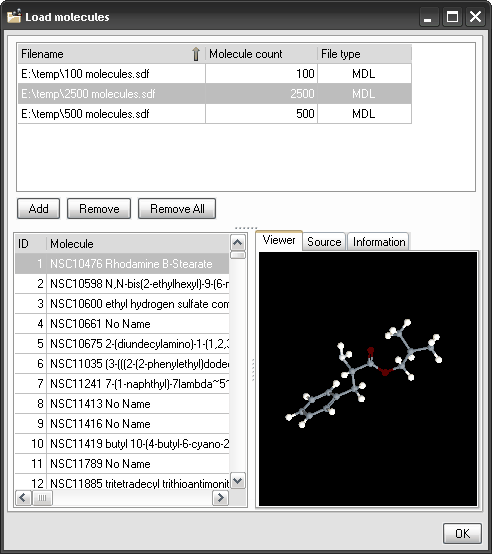
 Input files.
Together with the molecule formats allowed in the previous versions of
Dragon (MDL, Sybyl, HyperChem, Macromodel and Smiles), new formats are
now supported: CML (Chemical Markup Language) and HyperChem format as a
unique file. There are no restrictions on the number of molecules and
on the atom types. The novel Dragon graphical interface for molecule
import is very flexible allowing also selection of structures that are
stored by different file formats and located in different folders.
Molecular structures stored in files of different types can be loaded
and contemporarily processed in the same batch. Input files.
Together with the molecule formats allowed in the previous versions of
Dragon (MDL, Sybyl, HyperChem, Macromodel and Smiles), new formats are
now supported: CML (Chemical Markup Language) and HyperChem format as a
unique file. There are no restrictions on the number of molecules and
on the atom types. The novel Dragon graphical interface for molecule
import is very flexible allowing also selection of structures that are
stored by different file formats and located in different folders.
Molecular structures stored in files of different types can be loaded
and contemporarily processed in the same batch.
Graphics and statistics.
Graphical tools are available in Dragon 6 for a preliminary descriptor
analysis. These graphs allow a preliminary analysis of molecule
distribution in the descriptor space, as well as a preliminary
correlation analysis. Look at the screenshots of Dragon 6.
A molecule viewer
is provided to display the molecular structures that have been imported
for descriptor calculation.
Loading of user-defined
variables. Dragon allows the user to import up to 200 variables
and string fields that can be added to the calculated molecular
descriptors such as experimental properties and information on the
processed molecules (e.g., CAS number, product code).
Extended utilities for
command line interface. The Dragon command line interface allows
the execution of Dragon as a background application. Dragon 6.0 has now
all the same facilities when running as command line interface as the
graphical interface. For instance, it is now possible to apply the pair
correlation criterion to exclude redundant descriptors from the output
file. Moreover, the novel script wizard that guides the user in
creating the script file can be directly launched by the graphical
interface.
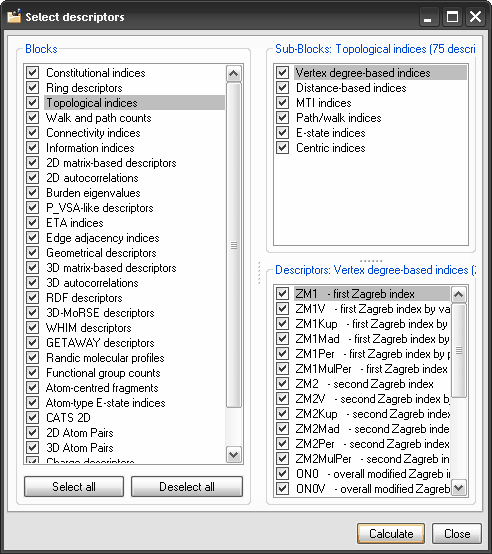
Descriptor calculation.
In Dragon 6.0, the user is allowed to customize descriptor calculation
and choose the atom weighting schemes before descriptor calculation,
whether apply logarithmic transformation to spectral moments and walk
and path counts. The novel window for descriptor selection allows also
selection of single molecular descriptors included in the different
blocks.
Extended capability to
process a variety of molecular structures. Restrictions on atom
types of molecules are now avoided. Therefore, Dragon is now able to
deal with any type of connected structure that also includes undefined
chemical elements.
Projects and templates.
Results of any calculation can be saved into a project that can be stored and
later reloaded to continue result analysis and export of different
selections of molecular descriptors. Moreover, to make recursive
calculation faster, a template can be created, where
information on descriptor selection and general options can be saved.
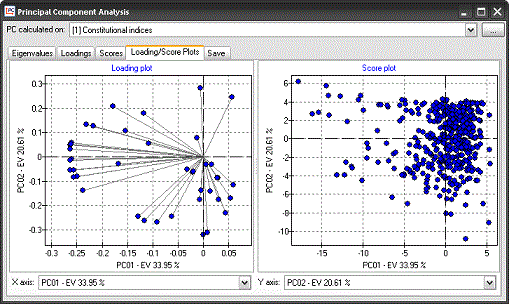
Principal Component
Analysis can be calculated on selected sets of descriptors. PCA
results can be easily exported or analysed with the graphical tools
provided in Dragon.
DProperties:
dProperties is an
easy-to-use application for the calculation of physicochemical
properties and drug-like indices. These properties can be used to
characterise molecules, for screening and filtering of molecule
databases.
dProperties calculates molecular
properties, such as constitutional features (molecular weight,
number of atoms, Hydrogen atoms, Carbon atoms, Nitrogen atoms, Oxygen
atoms, halogen atoms, sp3 hybridized Carbon atoms), unsaturation count
and index, hydrophilic factor, molar refractivity, topological polar
surface area, Moriguchi octanol-water partition coefficient,
Ghose-Crippen-Viswanadhan octanol-water partition coefficient, Surface
areas, McGowan volume, van der Waals volume, packing density index,
Verhaar base-line toxicities,
and drug-like indices (such as
Lipinski Alert Index, Drug-like scores and Lead-like Scores, Unsat
index, Dragon consensus drug-like score, property ranges).
|
Load
Molecules:
Select Descriptors:
Principle Component
Analysis:
|