Discovery Tools
From clustering and diversity analysis for chemical libraries to 2D and 3D molecular screening
|
| JKlustor |
|
|
JKlustor Summary
JKlustor - Clustering and diversity analysis for chemical libraries
JKlustor is a suite packed with
clustering methods, that performs similarity and structure based
clustering of compound libraries and focused sets - in both
hierarchical and non-hierarchical fashions. The suite also carries out
diversity calculations and library comparisons based on molecular
fingerprints and other descriptors. It is an essential tool in
combinatorial chemistry, virtual library design, and other areas where
large numbers of compounds are analyzed. |
 |
|
|
Features of JKlustor
Similarity based clustering
 Hierarchical method:
Hierarchical method:
Ward’s minimum variance method, speeded up with Murtagh’s
reciprocal nearest neighbour algorithm, creates tight and well
separated clusters. It's recommended to use it with smaller data sets,
like focused libraries with less than 100,000 structures. More on Ward clustering
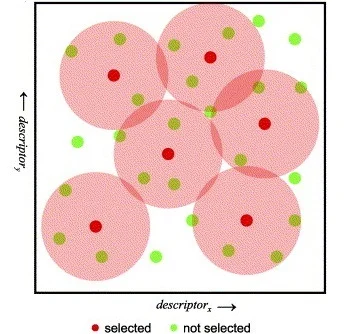
 Non-hierarchical method:
Non-hierarchical method:
The Sphere Exclusion clustering is based on fingerprints and/or other
numerical data, it can easily cope with millions of structures and it
is suitable for diverse subset selection. K-means cluster analysis
method aims to find the center of natural clusters in the input data in
a way that minimizes the variance within each cluster. Finally, the Jarvis-Patrick (Jarp) method uses a nearest neighbor approach and
performs variable-length clustering of chemical databases with hundreds
of thousands of structures contained.
|
|
Features of JKlustor
Similarity based clustering

Hierarchical methods:
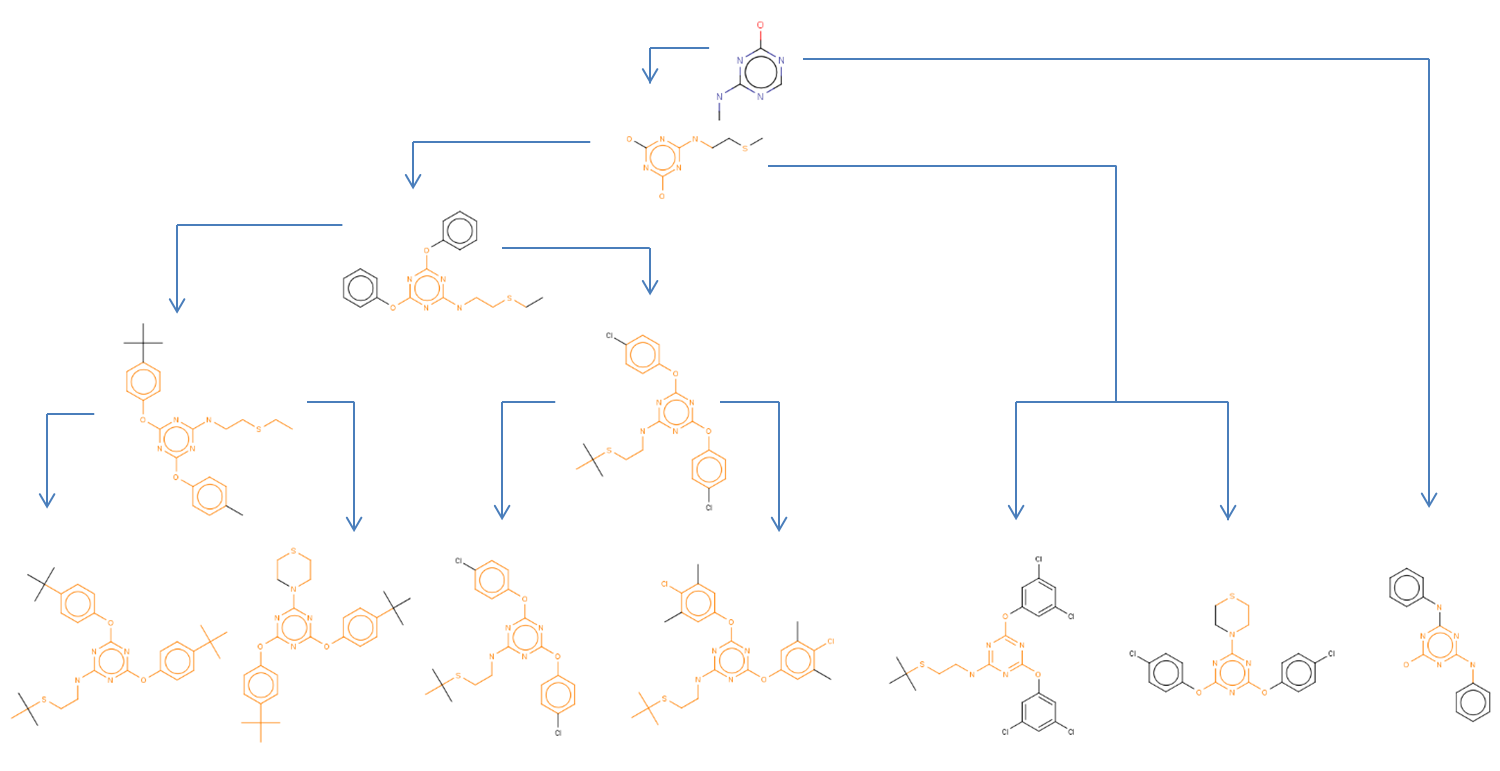
LibraryMCS identifies the largest substructure shared by several
molecular structures. It uses the hierarchical representation of
clusters (dendograms), and it vizualizes an alternative tree and table
view too. MCS profiling helps scientists explore screening results to
quickly identify novel scaffolds and new examples of active compound
families. The hierarchical SAR table enables viewing of clusters and
associated non-structural data. R-group decomposition can be also
performed using the MCS as the core structure for each cluster.
Non-hierarchical method:
JKlustor makes clustering available for pre-generated use Bemis-Murcko frameworks structures,
and therefore provides a convenient and quick way towards analyzing
large databases with millions of compounds.
|
 |
|
Features of JKlustor
Extending capabilities with descriptors and command line tools
JKlustor can use Chemaxon’s proprietary chemical and pharmacophore fingerprint technology, and also other user defined descriptors such as BCUT. For example, predicted or measured phys-chem properties (like pKa, logP etc.) can also help with clustering.
Command line tools in JKlustor:
- Diversity analysis - with Compr
command line it generates different types of similarity or
dissimilarity comparisons within a dataset (also see Diversity Set Selection) .
- GenerateMD - generates molecular descriptors for molecular stuctures.
- Jarp - clustering by a modified Jarvis-Patrick method.
- Ward - clustering by Ward's hierarchic clustering method using the RNN approach.
- LibMCS - Maximum Common Substructure based hierarchical clustering.
- CreateView - composing a new SDfile from an SDfile and a data table - It's useful for viewing the clustered results.
|
 |
|

|
JKlustor Benefits
Availability
JKlustor tools can be called upon from the command line or from the API
of JChem. JKlustor runs on many operating systems and can integrate
with many database engines. Full Java and .NET integration is
supported, as well as, connected to Oracle, MySQL, MS SQL Server, DB2,
PostgreSQL, Access, etc. databases. The LibMCS element comes with a
standalone GUI that allows users to browse/navigate through large sets
of data. Furthermore, maximum common edge sub-graph (MCES) and maximum
common substructure (MCS) clustering methods are also available as
Chemaxon components, for both, KNIME and Pipeline Pilot workflow
management systems.
|
|
| Screen Suite |
|

|
Screen Suite Summary
Screen Suite - Toolkit for 2D and 3D molecular screening
The Screen Suite is a ligand-based high throughput virtual screening
package that provides powerful tools for chemical (2D) and shape (3D)
similarity searches of large molecular libraries. The suite provides
several different sets of fingerprints, such as the Chemaxon chemical
fingerprint, pharmacophore fingerprint, ECFP/FCFP, dissimilarity
metrics and their optimization to tune your processes for optimal
search results. Commercially the suite is available as 2D Descriptor
package, Screen2D (including our chemical fingerprint) and Screen3D. It
is also available as a command line tool, from Java and .NET API and
workflow tools like KNIME or Pipeline Pilot. Screen Suite is a
progenitor of the novel MadFast similarity search engine, where
descriptor generation and similarity search using multiple metrics are
also available.
|
|
|
Screen Suite Features
Flexible descriptor accessibility in Screen Suite
The Screen2D Descriptor package contains fingerprint generators for
chemical structures, pharmacophores, extended connectivity (ECFP) and
functional class (FCFP) descriptors. It can be used to generate and
tune fingerprints for use in any screening process. Descriptor
generation is supported through Chemaxon’s Chemical Terms scripting
language, to create complex fingerprints which consider single or
combined molecular attributes that can be based on topological or
physicochemical properties like partition coefficients, hydrogen
bonding donor-acceptor accessibility or acidic properties.
|
 |
|
 |
Screen Suite Features
Diverse fingerprints
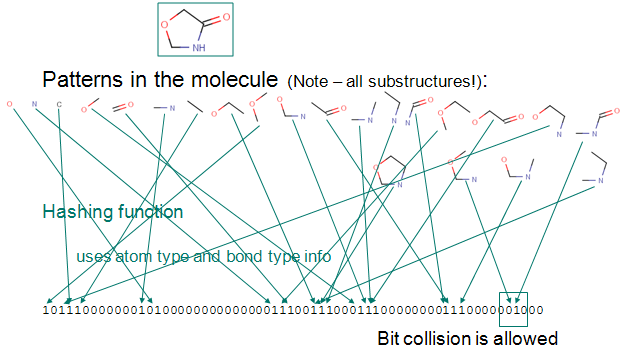
Chemaxon's screening technology heavily relies on the available chemical fingerprints and molecular descriptors:
- BCUT descriptors -
used in diversity analyses and QSAR; based on the connectivity table of
molecules with physchem properties like atomic charge, HBDA and
polarizability
- Chemical fingerprint -
used for structural searches in databases, similarity search or
diversity analysis of compound libraries; it is a path-based bit-string
descriptor describing molecular structures
- Pharmacophore fingerprints -
atom-based fingerprints characterizing binding related structural or
physchem properties that are thought to be responsible for
pharmacophore activity
- Circular fingerprints -
in contrast to path-based fingerprints ECFP/FCFP are not substructure
preserving descriptions, but are highly relevant for full structure and
similarity searches.
|
|
|
Screen Suite Features
Screen 2D
Screen 2D is a robust platform to perform
chemical similarity searches on large molecular databases. It offers a
fast process with up to 100,000 compounds per second being screened on
a single desktop PC. Modularity of the tool allows to use own
descriptor sets. Screen 2D comes with a set of metrics to measure
dissimilarity between compounds:
- Euclidean
- Tanimoto
- Tversky
- Dice
Screen 2D supports metrics optimization by
selecting appropriate training sets. The optimization step increases
enrichment significantly, giving smaller, more focused hit sets. It is
available through API and command line, via Pipeline Pilot and KNIME connectors, and some features are implemented within Instant JChem and JChem for Office desktop applications.
|
 |
|
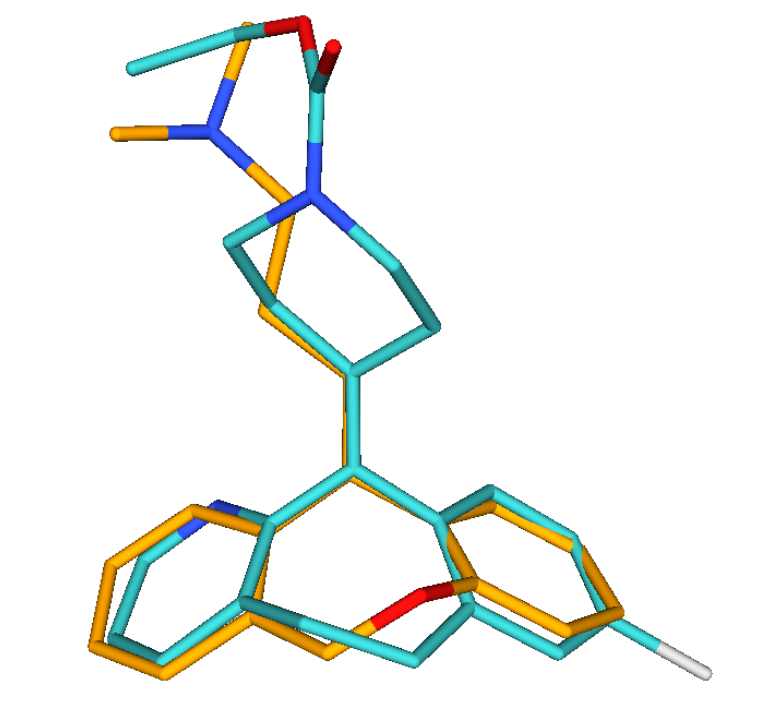 |
Screen Suite Features
Screen 3D
Screen Suite includes 3D shape similarity based searching in addition
to topology based screening. It introduces a novel flexible alignment
technique, which provides reasonable alternative hit results as
compared to previous 3D screen engines. The key feature of Screen 3D is
definitely its high speed, however, it also compares very well in
providing better enrichment in the first 1% of the hits than most
alternative software. Scalability of the tool makes it suitable to
search in very large molecular databases.
Screen 3D can be used to carry out different shape similarity searches.
Beyond fully flexible matching it can align flexible database compounds
to rigid query molecules (rigid-flexible). Alternatively it is capable
of aligning rigid database compounds to rigid query molecules
(rigid-rigid) using on-the-fly generated conformation ensembles.
Benefits of Screen3D can also be exploited as a plugin in Design Hub
idea assessment and management platform. |
|
Other Products:
|
|
|
Marvin know More |
Chemical Naming know More |
|
| Markush Technology know More |
Chemical Structure Representation know More |
|
| Chemicalize know More |
Compound Registration know More |
|
| Reactor know More |
JChem for Office know More |
|
| Design Hub know More |
JChem Engines know More |
|
| Calculators and Predictors know More |
Compliance Checker know More |
|
| Instant JChem know More |
ChemCurator know More |
|
| MarvinSketch & Marvin JS know More |
|
|